细胞死亡是多细胞生物中一种生理或病理现象,它用来消除多余或有害细胞,对于多细胞生物的发育、正常细胞的更新以及各组织的正常结构和功能维持具有重要意义。细胞死亡可以通过多种方式发生,其中包括细胞凋亡、焦亡、铁死亡、铜死亡、自噬以及相关细胞死亡等过程。本文将简要介绍这些细胞死亡形式的基本机制,并总结它们对应的标志物。
细胞凋亡(Apoptosis)是指为维持内环境稳态,由凋亡小体和Caspases介导的细胞程序性死亡过程。目前,细胞凋亡分为三种:
(1)线粒体凋亡途径:在细胞受到外部/内部刺激后,线粒体渗透转换孔打开,导致内部的促凋亡蛋白-细胞色素C和Apaf-1释放到细胞浆中,细胞色素C通过Apaf-1的CARD结构域与Caspase-9结合形成凋亡复合物,诱导Caspase-9 活化,进而活化Caspase-3,引发凋亡过程。
(2)死亡受体途径:死亡配体(FasL)与死亡受体(Fas)结合后,通过特殊死亡结构域(DD)招募FADD,FADD由DD和DED两个结构域构成,其DD结构域可以结合到Fas,而其DED结构域可以与Caspase-8上的DED结构域互补结合,从而形成FASL/Fas/DED/Caspase 8复合物,即死亡诱导信号复合物(DISC),DISC中的Caspase-8酶原发生自身切割来获得活性,活化的Caspase-8切割Caspase-3酶原,使其活化,引发凋亡过程。
(3)内质网应激途径:在内质网应激时,Ca2+从内质网中释放入细胞质后会激活内质网附近的钙调蛋白分解酶(Calpain),它可以作用于Caspase-12使之活化并释放入细胞质,诱发细胞凋亡。同时Ca2+可以通过激活Ca2+/钙联蛋白调节的钙调神经磷酸酶(Calcineurin),使得前凋亡蛋白Bad去磷酸化,激发细胞色素C的释放,诱导凋亡。
标志物解析
(1)Bax/Bcl-2:Bax是促凋亡基因,其可被p53诱导表达,对肿瘤有抑制作用。在细胞凋亡时表达增加。Bcl-2是凋亡抑制基因,其编码的蛋白具有抑制细胞凋亡的作用,且不影响细胞增殖。Bcl-2与Bax是同源基因,两者共同构成了细胞凋亡的微调变阻器。因此以两者的比值作为凋亡的指示是有一定意义的。
(2)Caspase-3/8:Caspase系统的激活对于细胞凋亡的发生至关重要,因此Caspase家族蛋白在细胞凋亡时表达均增加。
细胞自噬(Autophage):细胞自噬是指双层膜(自噬泡)包裹部分胞质和细胞内需要降解的细胞器、蛋白质等形成自噬体(Autophagosome),最后与溶酶体融合形成自噬溶酶体(Autophagolysosome),降解其所包裹的内容物,以实现选择性地清除自身受损、衰老或过剩的生物大分子和细胞器,释放出游离小分子供细胞回收利用的正常动态生命过程。自噬通常被认为是一种生存机制,参与调节细胞内物质合成、降解和重新利用之间的代谢平衡。
目前,自噬分为三种类型:
(1)巨自噬(Macroautophagy):通过形成具有双层膜结构的自噬体(Autophagosome)包裹胞内物质,最终自噬体与溶酶体融合进行降解。
(2)微自噬(Microautophagy):通过溶酶体或液泡发生膜内陷直接吞没特定的细胞器进行降解。
(3)分子伴侣介导的自噬(Chaperone-mediated autophagy, CMA):具有KEFRQ样基序的蛋白在HSP70分子伴侣的帮助下,将蛋白质由折叠的状态恢复为未折叠的状态,然后通过LAMP-2A转运体转运到溶酶体内进行降解。
标志物解析
(1)LC3B为经典的自噬标志物,LC3B在C端被蛋白酶切割后会形成游离型的LC3B-I型(16kDa),而在自噬发生后LC3B-Ⅰ会与磷脂酰丝氨酸结合形成膜型LC3B-II型(14kDa),其为自噬体膜上的结构蛋白。因此在自噬发生时LC3B-II会增加,且LC3B-II/LC3B-I的比值也随之增加。
(2)Beclin-1为自噬体形成过程中的一个必需分子,其能够介导其他自噬蛋白定位于吞噬泡,从而调节自噬体的形成与成熟,在自噬中表达增加。
(3)p62是一种泛素结合蛋白,是反映自噬活性的标记蛋白之一。在自噬过程中,p62 与泛素化的蛋白质结合,再与定位于自噬小体内膜上的 LC3-II 蛋白形成复合物,共同在自噬溶酶体内降解。当自噬活性减弱、自噬功能缺陷时,p62 蛋白会在细胞质中不断累积。p62的含量间接反映自噬小体清除水平,在细胞发生自噬时表达降低。
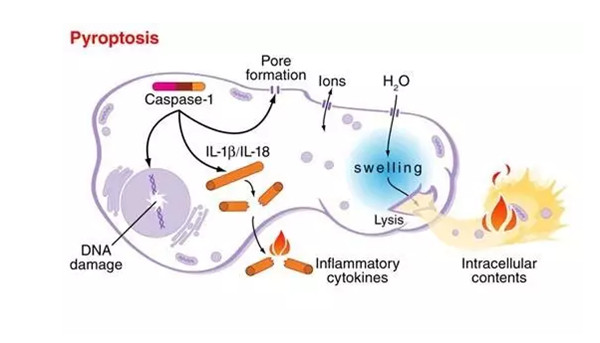
细胞焦亡(Pyroptosis):一种由GASD家族蛋白执行的炎性小体依赖的程序性死亡过程,其比细胞凋亡发生的更快。细胞焦亡通常伴随着大量促炎症因子的释放,是依靠Caspase-1形成质膜孔,导致促炎细胞因子(如IL-1β和IL-18)的释放和细胞裂解。细胞焦亡一种对抗细胞内病原体的重要先天免疫效应机制,广泛参与感染性疾病、神经系统相关疾病和动脉粥样硬化性疾病等的发生和发展。
目前,细胞焦亡的类型分为两种:
(1)经典通路:由炎性小体组装介导并伴随GSDMD裂解和IL-1β和IL-18释放。炎症小体是多分子复合体,在胞质模式识别受体(PRRs),识别病原体上的病原体相关的模式分子(PAMPs)与危险相关的模式分子(DAMPs)后被激活,从而促进下游信号通路,导致I型干扰素的产生和促炎细胞因子的释放,并与Caspase-1和ASC组装形成炎性小体导致细胞焦亡。
(2)非典型通路:在非典型的焦亡途径中,上游Caspase-4/5复合物缺失,Caspase蛋白会通过N端CARD直接结合细胞内脂多糖(LPS)而被激活。活化的Caspase-4/5/11将GSDMD切割为N-GSDMD,N-GSDMD发生寡聚化并转移到细胞膜上形成质膜孔,导致K+外排,诱导炎性小体组装,最终导致焦亡。
标志物解析
Garsdermin(GSDM)家族蛋白均对细胞焦亡的产生有重要影响,因此,该家族成员均可作为发生细胞焦亡的标志物(GarsderminA/B/C/D/E),其中以GSDMD和GSDME作为细胞焦亡标志物居多。因此在检测细胞焦亡标志物时,应该在未诱导细胞焦亡的情况下检测到全长的GSDM(55kDa左右),而在诱导细胞发生焦亡后,GSDM被水解为两部分,即PFD(35kDa左右)和RD(25kDa左右)。
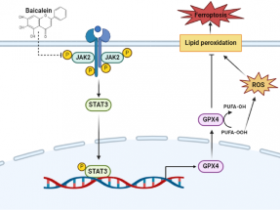
细胞铁死亡(Ferroptosis):是一种铁依赖性脂质过氧化物的积累所导致新型细胞死亡的方式。主要是由铁超载和活性氧(ROS)依赖的脂质过氧化物累积引起的。线粒体是铁利用、分解代谢和合成代谢途径的主要细胞器,细胞发生铁死亡时线粒体与正常线粒体相比膜密度更致密,体积更小,嵴减少或消失,外膜破裂等。
标志物解析
(1)GPX4:GPX4(22kDa)为细胞铁死亡的关键调控因子,GPX4可利用其催化活性,削弱脂质过氧化物毒性,维持膜脂质双分子层稳态,从而抑制铁死亡的发生。而用GPX4抑制剂RSL3处理后,RSL3与GPX4共价结合而使其GPX4失活,从而导致细胞内过氧化物堆积,引发铁死亡。
(2)FTH1:FTH1(21kDa)会破坏铁自噬体,从而抑制铁死亡。
(3)ACSL4:ACSL4(79kDa)是调节脂质组成的关键酶,可促进铁死亡。
(4)PTGS2:PTGS2(69kDa)在小鼠铁死亡细胞中表达水平明显提升,但是PTGS2的抑制剂并不能影响铁死亡,所以PTGS2只可能是铁死亡的标志物而不是推动铁死亡的功能分子。
细胞铜死亡(Cuproptosis)是一种新发现的细胞死亡现象,即铜离子稳态失衡诱导细胞发生的死亡过程。铜死亡是通过铜与三羧酸循环(Tricarboxylic acid cycle,TCA cycle)的脂化组分直接结合发生的。这导致脂酰化蛋白聚集和随后的铁硫蛋白损失,使得蛋白毒性应激并最终导致细胞死亡。
线粒体中的铁氧还原蛋白(Ferredoxin 1, FDX1)是铜死亡发生的核心分子,FDX1可以将Cu2+还原为毒性更强的Cu+,同时FDX1还可以使丙酮酸脱氢酶复合体中的DLAT和DLST及硫辛酰化相关酶LIAS发生脂酰化,从而赋予其酶活性。在细胞内铜离子发生积累时,FDX1可以将Cu2+还原为Cu+,并催化DLAT、DLST和LIST发生脂酰化,而Cu+进一步结合到DLAT的脂酰化部位,使DLAT发生寡聚化,从而获得铜毒性。此外,Cu+稳态失衡会导致细胞内铁硫蛋白丢失,HSP70表达水平增加,引发蛋白质毒性应激。以上两条途径共同造成了细胞铜死亡的发生。
标志物解析
(1)FDX1为一种铁氧还原蛋白,是铜死亡发生的核心分子,一方面FDX1可以将Cu2+还原为毒性更强的Cu+来诱发铜死亡;另一方面可以催化丙酮酸脱氢酶核心结构蛋白发生脂酰化,通过影响三羧酸循环的发生来影响细胞发生铜死亡。
(2)LIAS为硫辛酸合成酶,可作为FDX1的脂酰化底物参铜死亡的发生。
(3)HSP70为热休克蛋白,在铜死亡中引发蛋白质毒性应激反应。