实验室常用的动植物组织和细胞RNA提取方法包括传统的含氯仿的RNA提取试剂和无氯仿RNA提取试剂。这两种方法的基本原理是利用单相裂解试剂进行RNA提取。在这个过程中,生物样品被异硫氰酸胍和苯酚的单相溶液裂解,然后加入氯仿(或溴氯丙烷),混匀后离心,使混合物分为水相和有机相。RNA主要存在于水相中,而DNA位于水相和有机相的分界处,变性蛋白则存在于有机相中。
分离后的水相可以通过异丙醇沉淀来回收RNA。如果需要提取DNA和蛋白质,可以使用顺序沉淀分离方法:DNA可以通过乙醇沉淀从中间相中获得,蛋白质则可以通过异丙醇沉淀从有机相中获得。
回收的DNA片段大小约为20kb,适用于PCR模板。然而,由于暴露于胍盐,蛋白质会变性,因此主要用于免疫印迹实验。这些方法为实验室提取RNA、DNA和蛋白质提供了可靠的手段,有助于进行分子生物学研究和实验工作。
实验操作实例
接下来我将以果蝇成虫为例详细介绍两种方法的步骤
样本处理:
A. 组织样本
1.将新鲜组织用液氮速冻,迅速转移至液氮预冷的研钵中,用研杵研磨,期间不断加入液氮,直至研磨成粉末状(无明显可见颗粒)。
▲研磨不彻底会影响RNA的得率和质量。
2. 将研磨成粉的样品转移至离心管中,大约每50 mg组织加入1mL 含氯仿 (500μL 无氯仿试剂), 涡旋振荡至充分裂解,室温静置5 min。
▲每500μL 无氯仿试剂最多可裂解50 mg动物/植物组织,过多的样本量可能会导致裂解不充分,并使产物纯度降低。肝脏、脾脏、肾脏等组织DNA/RNA含量丰富,过多样本量还会导致gDNA残留或产量降低。
▲若没有条件用液氮研磨,可将新鲜组织尽量剪碎浸泡在单相裂解试剂中,用电动匀浆器高速匀浆至组织块彻底裂解(这里我使用手动研杵研碎,手动研磨时应注意避免局部积热,尽量在冰上进行)。
3. (可选)11,200 rpm(12,000 x g)室温离心5 min。小心吸取上清至新的1.5 ml离心管,切勿吸取沉淀。
▲若样本含有较多蛋白质、脂肪、多糖或肌纤维、植物块茎等,可离心去除不溶物质,离心得到的沉淀包括细胞外膜、多糖、高分子量DNA,RNA存在于上清中。提取脂肪含量较高的组织样本时,若上层含有大量油脂,应去除。转移上清进行后续操作。
B. 悬浮细胞样本
1.离心收集细胞,弃尽上清,每1×105~1×107个细胞加入1mL 含氯仿 (500μL 无氯仿试剂)。
2.涡旋振荡或用移液器反复吹打直至充分裂解,室温静置5 min。
▲对于冻存细胞,加入无氯仿试剂应立即进行振荡,否则会导致裂解不充分。
C. 贴壁细胞样本
1.弃去细胞培养液,用1×PBS清洗一次,弃尽废液。
2.常规6孔板每孔或直径3.5 cm平皿(约10 cm2培养面积)的细胞加入500μL 无氯仿试剂,每100mm平皿加入1mL 含氯仿,使之充分覆盖到细胞表面,然后用移液器反复吹打细胞使其脱落。
▲对于贴壁牢固的细胞(细胞团)可采用细胞刮或者洁净的枪头剥离,或增加单相裂解试剂的用量,亦可在加入单相裂解试剂之前采用胰酶将细胞消化下来,然后按照悬浮细胞处理。
3.将裂解液转移至1.5 ml离心管,涡旋振荡或用移液器反复吹打直至充分裂解,室温静置5 min。
▲浸在含氯仿试剂中的样本可以在-60~-80°C存放至少一个月

含氯仿法提取RNA
1. 孵育匀浆样品,室温5min,以完全裂解核蛋白复合物;
2. 每毫升含氯仿加入0.2mL氯仿;盖紧管盖用力摇动15s,并在室温下孵育2-3min;
▲不要涡旋样品,因为它会导致DNA污染。
▲注意使溶液充分混匀为均一溶液,混匀不彻底影响RNA提取效率和杂质去除效率。
3. 4°C,12000g 离心15min,混合物分为三相,RNA仅存于上层水相;
4. 小心地将上层水相转移至新Ep管内,注意不要扰动中间层;
▲每1mL 含氯仿可获得约600μL水相,为防止DNA污染推荐只回收500μL。
5. 每毫升含氯仿加入0.5mL异丙醇沉淀RNA,颠倒混匀,室温孵育10min;
▲注意使溶液充分混匀为均一溶液,混匀不彻底将无法使RNA沉淀。
▲低浓度样品中回收RNA时效率很低,可以在此步加入1μL糖原(20mg/mL)。
▲为防止温度过高使RNA降解,也可以置于冰上、-20°C冰箱中进行沉淀。
6. 12000g离心10min,收集RNA沉淀;
7. 弃上清,并用吸头完全去除残留液体;
8. 用75%乙醇洗涤一次RNA沉淀,每毫升含氯仿加入至少1mL 75%乙醇,涡旋混合样品液,然后在4°C 7500g离心5min;重复清洗一次;
▲沉淀于75%乙醇的RNA可储存在2~8°C至少一周,-5~-80°C至少一年。
9. 完全去除乙醇,空气干燥或真空干燥RNA沉淀10min;
▲除去乙醇后,沉淀应该是半透明的,在干燥时,沉淀将变成透明凝胶体状。不要真空离心干燥的RNA。重要的是不要让RNA沉淀完全干燥,因为这将大大降低其溶解度。
10. 用20~100μL的RNase free水溶解RNA,反复吹打几次。
▲部分方案中建议在55-60°C温育10min,这里我个人不建议进行加热;若RNA过于干燥,可在40°C温育直至其完全溶解。
无氯仿法提取RNA
1.向孵育匀浆样品中加入Dilution Buffer。(动植物组织:每500μL 无氯仿试剂加入100μL Dilution Buffer;细胞:每500μL 无氯仿 Reagen加入150μL Dilution Buffer。)盖紧离心管盖,涡旋振荡至溶液充分混匀,室温静置5 min。
▲振荡时注意使溶液充分混匀为均一溶液,混匀不彻底影响RNA提取效率和杂质去除效率。
2. 12,000g室温离心15 min。
3.小心取出离心管。此时样本中蛋白质、DNA、多糖等杂质沉淀到管底,RNA分布在上层溶液中,小心吸取上清溶液(约500μL)至一个新的离心管中。
▲上层溶液的体积约占总体积的90%,用500μL 无氯仿试剂进行提取,上层溶液约为550μL ;吸到下层沉淀会导致基因组及杂质污染。
▲部分样本上清轻微浑浊或有颜色,可继续后续操作,不影响产量和纯度。
▲组织投入量为50 mg时,吸取上清溶液体积建议减少至450μL ,防止吸到下层沉淀。
4.在得到的上清溶液中加入等体积异丙醇,上下颠倒充分混匀,室温静置10 min。
5. 12,000g室温离心10 min,离心后在管侧和管底可以看见白色凝胶状沉淀,小心弃去上清,勿丢失沉淀。
▲RNA含量少会导致沉淀不明显,小心弃去上清,勿丢失沉淀。
▲部分特殊样本如:水稻叶RNA沉淀会分散在离心管壁上导致白色沉淀不明显,小心弃去上清,步骤7离心后沉淀可见。
▲为减少杂质残留,此步骤尽可能将上清弃干净,勿丢失沉淀。
6.加入1 ml 75%乙醇。轻弹管底,让沉淀悬浮起来,并上下颠倒数次。
7. 8,000 g室温离心3 min,弃去上清,勿丢失沉淀。
8.重复步骤6和7一遍,弃尽上清。
▲为减少杂质残留,应尽可能的将上清液弃干净。建议弃去大部分上清后,短暂离心将所有液体甩至管底,再用移液器将剩余液体吸掉,注意不要吸到沉淀。
9.室温放置晾干,加入20- 100μL RNase-free ddH2O溶解沉淀,室温涡旋3 min(或使用移液器反复吹打),使RNA沉淀充分溶解。提取的RNA产物可以分装后在-85 ~ -65°C长期保存,在-30 ~-15°C短期保存。
▲通常,RNA沉淀晾干2 - 3 min即可,不要过度晾干,RNA完全干燥后会很难溶解。
▲RNA产物需要充分溶解,否则可能导致浓度定量失准。
使用超微量紫外分光光度计,测量提取RNA样品的A230、A260、A280,用来计算RNA样品的浓度、估计RNA纯度。
计算溶液中RNA浓度的公式:NA样品浓度(μg/mL)= A260×40×稀释倍数
A260不能区分RNA和DNA,因此,建议用无RNase的DNase处理RNA样品去除DNA污染。 此外,污染物如苯酚和蛋白质会影A260的读数; 苯酚在波长为260nm处有强烈的吸收,而芳香族氨基酸的吸收在280nm处。 在260nm处吸光度相对于280nm的吸光度比值( A260/A280)可以用来评估RNA样品中蛋白质的污染水平。纯的RNA的A260/A=值接近2.0。
如果有明显的蛋白质或酚污染,A260/A280 比小于2.0,用紫外分光光度法将不可能准确定量核酸含量。 另一个比值A260/A230可用于评估有机化合物或高离液盐( chaotropic salt)的污染水平,它们在波长230nm处有强烈吸收,导致低A260/A230。 理想情况下,提取RNA的A260/A230的比例应接近2.0。
▲A260/A280 比值依赖于pH和离子强度。随着pH的增加,A280 降低, 但A260 不受影响,从而A260/A280的比值增加。因为纯水pH为酸性,如果使用它们溶解RNA, A260/A280的比值可能偏低。因此为了准确的可重复的读数,最好使用缓冲液如TE (pH 8.0)作为稀释剂和空白对照。
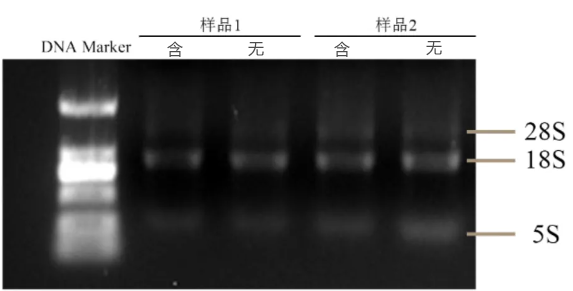
琼脂糖凝胶电泳分析:
提取的RNA质量优劣可以通过简单的非变性琼脂糖凝胶电泳进行检测,使用MOPS或TBE缓冲液、配制0.8%琼脂糖凝胶,在4~5V/cm电压下进行电泳,若RNA纯度较好将会有清晰的三(四)条带(28S、18S、5.8/5S)且亮度均一(上样质量一样)。
常见问题&解决方案
| 常见问题 | 原因 | 解决方案 |
| RNA产物溶解不完全 | 1.干燥时间过长 | 75%乙醇清洗后控制干燥时间,避免过分干燥;延长溶解时间或55~60℃水浴加热2~3 min |
| 2.产物量过多 | 增加ddH2O投人量,延长溶解时间或55~60℃水浴加热2~3min | |
| 3.杂质过多 | 样本充分裂解后,离心取上清后再进行后续操作 | |
| RNA降解 | 1.存在RNase污染 | 确保所有离心管、枪头及相关溶液都必须无RNase污染;做好防护工作,戴口罩和一次性干净手套,并在单独的洁净区操作 |
| 2.样本保存不当或 样本保存时间过久 |
采用新鲜样本或经液氮速冻后保存于-85~-65°C的样本 | |
| 3.样本反复冻融 | 样本保存时,建议分装后保存,避免因反复冻融导致的降解;从液氮中取出的样本应迅速加入裂解液并充分混合均匀,防止样本因置于室温时间过长或与裂解液混合不均匀导致RNA降解 | |
| 4.电泳原因 | 电泳前将电泳槽用3%双氧水浸泡20min,或使用RNA酶和核酸清除剂处理5min,然后用RNase-free ddH2O进行冲洗;电泳缓冲液用RNase-free ddH2O配制;更换新的Loading Buffer | |
| 抑制下游或纯度低 | 1.蛋白质污染 | 减少样本起始投入量;增加单相裂解试剂体积 |
| 2.多糖污染 | 减少样本起始投入量;增加样本处理A3步骤 | |
| 3.脂肪污染 | 减少样本起始投人量;增加样本处理A3步骤 | |
| 4.盐残留 | 增加75%乙醇漂洗次数 | |
| 基因组DNA污染 | 1.不小心吸取到中 间层(TRIzol法) |
离心结束减少晃动,减少吸取上清量 |
| 2.样本投入量过高 | 减少样本起始投入量 | |
| 增加单相裂解试剂体积 | ||
| 样本裂解时,加适量HAc | ||
| 逆转录时选择含有基因组去除模块的逆转录试剂 | ||
| 设计引物时选用跨内含子的引物,从而避免基因组DNA模板参与扩增反应 | ||
| 加入异丙醇离心后看不见沉淀 | 1.样本投入量低或 RNA含量低 |
加入异丙醇后在2~8℃或-30~-15C放置10-30min后再离心;加入1μL糖原(20mg/mL)进行共沉淀 |
| 2.沉淀丢失 | 进行弃上清操作时,采用吸取而不是倾倒的方法,勿丢失 沉淀 |
|
| 3.样本自身代谢产物多 | 沉淀会分散在离心管壁上,弃上清时沿液面缓慢吸取 |
更多详情产品查看:RNA提取相关产品-艾美捷科技有限公司