
在前面的《啥?真的有“消瘦基因”》一文中,我们给大家介绍了现在有科学家发现了与极度消瘦有关的基因组区域。今天我们再来聊聊与之相对的肥胖,嗯,没错,科学家们也发现了与肥胖相关的基因,惊不惊喜,意不意外?

肥胖症是一组常见的代谢症群。尽管普遍认为热量摄入和能量消耗之间的失衡是导致肥胖的重要原因,但是多年来积累的数据表明,造成一些人比其他人更加肥胖的原因没有这么简单。首先来说个很意外的发现:嗅觉与体重增加速度有关,嗅觉好的人更容易胖……,太恐怖了,啥时候鼻子灵还有坏处了,让那些靠鼻子吃饭的调香水的人情何以堪?事情是这样:来自美国加州大学伯克利分校的研究团队用小鼠做了这样一个实验,让嗅觉正常与嗅觉失灵的小鼠进食等量的食物。结果发现,嗅觉正常的小鼠体重增加速度明显大于嗅觉失灵的小鼠。该研究于2017年7月5日发表在Cell Metabolism上。

实验中两鼠的体型对比,左边嗅觉正常,右边嗅觉失灵。
以往,人们总认为嗅觉只是影响食欲而已。随着年龄的增长或受疾病影响,我们的嗅觉下降,食欲降低,体重自然而然会减轻。反之,嗅觉好的人食欲更强,吃得更多,自然会胖啦!但该研究首次证实,即便食量不变,嗅觉也能直接影响人体的能量代谢方式。换句话说,体重增减不是简单的卡路里加减法。打个简单的比方,在聚会上大家都吃一样多的一块蛋糕,可能只有觉得蛋糕特别香的小伙伴贴膘最多。
科学家认为,如果什么也闻不到,我们的身体则更倾向于燃烧摄入的能量而非将它们储存起来。其原因或许是,我们在饥饿状态时的嗅觉灵敏度明显高于饱腹状态。因此,嗅觉失灵时,身体很可能以为自己已经吃饱了,便不再积极存储脂肪。嗯,听着有点道理,让我想起以前看到的关于火鸡妈妈的传说。据说火鸡妈妈保护小火鸡的法则简单粗暴:攻击身边的一切会移动的物体,除非它能发出小火鸡的叫声。这在一般情况下没毛病,但如果有一天火鸡妈妈聋了,悲剧就发生了,火鸡妈妈啄死了所有小火鸡,虽然它们不仅看起来像小火鸡,而且充满信任的朝妈妈跑过来。呜呜呜。。。可能我们的身体也是用非常简单的法则在调节脂肪的储备。
为了验证自己的想法,实验中,研究人员在保留成年小鼠鼻腔内干细胞的前提下,破坏了嗅神经,使小鼠暂时失去了嗅觉。结果发现,嗅觉失灵的小鼠其体内的米色脂肪细胞(皮下存储脂肪的细胞)渐渐转变成褐色脂肪细胞(燃烧脂肪酸产生能量的细胞),此外,白色脂肪细胞(内脏周围存储脂肪的细胞)数量也大大减少。然而,代价是小鼠体内的去肾上腺激素水平也大幅增高。要知道,人体内该激素水平持续升高则极有可能诱发心脏病。如此看来,通过破坏嗅觉来减重的方法还是太激进了些。对此,研究人员表示,类似的方法更适用于治疗严重肥胖的患者,并在其代谢系统基本正常之后即时恢复嗅觉,以保证体内各激素的正常分泌。
言归正传,啥是肥胖基因?肥胖基因,是指基因所编码蛋白质是一种食欲与能量平衡调节途径的组成部分,而这种途径的失衡直接或者间接导致体脂肪的积累和体重增加。当第一个隐藏在肥胖症背后的关键基因被发现后,科学家们无不感到欢欣鼓舞。然而没有人--甚至包括发现这种基因的糖尿病专家--知道这种名为FTO的基因到底是如何工作的。
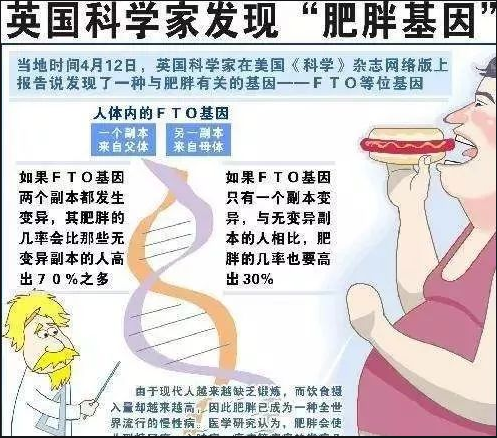
后来,一个研究小组向着破译FTO蛋白质让人发胖的原因迈出了关键一步。FTO的发现源于对2型糖尿病相关基因的搜索,但研究人员很快便意识到,它所扮演的真正角色与肥胖症直接相关。尽管科学家一直认为遗传因素可能是造成肥胖症的重要原因,但相关基因的敲定却困难重重。在这项发表在《科学》杂志上的研究中,研究人员对大约39000人进行了采样分析,断定携带一种FTO变异的两个副本的人,其体重增加了约3公斤,这是关于一种常见肥胖症基因的第一个清晰例证。然而随后的研究工作却使科学家遭遇了瓶颈,即FTO究竟干了什么以及它的运行机制到底是什么?
为了回答这些问题,由几家英国研究机构组成的科研小组分别对FTO进行了不同侧重的研究。其中剑桥大学的遗传学家Stephen O'Rahilly和他的同事尝试了解FTO蛋白质对于动物是如何作用的。研究人员在实验室小鼠体内发现,其大脑下丘脑--用来帮助调节机体的能量平衡--中的FTO含量非常高。与对照组的小鼠相比,下丘脑中FTO减少60%的小鼠往往会拒绝进食。这意味着,FTO在食欲的控制中可能扮演了一个重要角色。而牛津大学的科学家则将这项研究又向前推进了一步--对FTO蛋白质的基因序列进行了研究。
结果显示,这种蛋白质的作用更接近于酶,能够通过去除一个甲基修复脱氧核糖核酸(DNA)。更多的研究证明,FTO蛋白质能够使DNA脱甲基化。但是这项研究的主持人之一、牛津大学的酶学专家Christopher Schofield承认,依然有一些缺失的环节没有搞清--被FTO酶脱甲基的到底是什么基因或基因组,以及这一过程如何对肥胖症造成影响。英国埃克塞特市半岛医学院的Andrew Hattersley认为:"这项研究迈出了至关重要的第一步。"Hattersley参与了FTO基因的最初发现工作。但他与Schofield都承认,研究并没有就此结束,FTO如何改变体重以及是否真的通过食欲起作用都是科学家下一步的工作方向。一种缺少FTO的转基因小鼠将帮助解答这一难题,而这种啮齿动物的培育工作正是研究人员目前迫切需要解决的问题。科学家已克隆出了5 个与人的食欲及体重调节有关的基因,即OB基因、LEPR基因、PC1 基因、POMC基因和MC4R基因。
一、人的OB基因定位于第7号染色体长臂(7q31.3),在人类基因组中为单拷贝,全长约20kb,含有3个外显子,外显子全长4240bp 。OB基因只在脂肪组织中表达,其编码产物瘦蛋白是一种分泌性蛋白,即瘦素(Leptin),或瘦蛋白,是由167个氨基酸残基组成的。瘦素在脂肪组织合成后,分泌到血液中,在血液中与其受体LEPRe结合。
二、人的瘦素受体基因LEPR 定位于第1号染色体短臂(1p31),其编码产物瘦蛋白受体属于类细胞因子受体家族,共有6 种,即Ra ,Rb ,Rc ,Rd ,Re和Rf ,它们是LEPR 基因转录后通过不同剪切而生成。这些受体广泛分布于脑、心、肝、肾、肺、脾、胰脏、睾丸和脂肪组织中。瘦蛋白与受体LEPRe 结合后生成瘦蛋白2Re;后者将瘦素带入脉络膜,在此处瘦素与LEPRa 结合,生成瘦素2Ra;瘦素2Ra将瘦素输送到脑脊液,在这里瘦素与广泛分布在下丘脑的LEPRb 结合,生成瘦素2Rb。LEPRb 是瘦素各种受体中唯一的具有信号传道作用的跨膜蛋白,它在下丘脑产生的生理效应之一是诱发下丘脑神经细胞POMC 基因表达加强。
三、人POMC 基因定位于人类第2 号染色体短臂(2p23.3),其编码的蛋白质是一种前激素原。该蛋白质在前转变素酶1(proconvertase1,PC1) 的作用下分解成促肾上腺皮质激素(adrenocorticotropic hormone,ACTH)和促黑素细胞激素(α-melaocyte-stimulating hormone,α-MSH),后者在下丘脑与黑皮素4 受体(melanocortin 4 receptor ,MC4R))结合。
四、人PC1 基因定位于第5 号染色体长臂(5q152 21),全长3.3kb ,其编码产物是一种含有753个残基的蛋白酶,在神经内分泌组织中特异性表达,属于丝氨酸蛋白酶家族,其功能是将激素原(pro-hormone) 转化为激素,因此称为激素原转化酶(pro-hormone convertase,PC1) 。
五、人MC4R 基因定位于基因组第18 号染色体长臂(18q22),该基因主要在下丘脑神经细胞中表达,是瘦素介导的食欲调节途径中最末端的基因,由阿黑皮素原(POMC)衍生的α-MSH 在下丘脑与其受体MC4R 结合,产生包括调节食欲在内的生理效应。
当身体脂肪含量增加时,瘦素合成分泌增多;瘦素与其受体结合生成瘦素-Rb,后者在下丘脑诱发神经细胞POMC基因表达加强;POMC的强表达导致其分解产物α-MSH 的浓度升高;α-MSH 进而与其受体MC4R 结合,产生抑制食欲的生理效应。食欲被抑制导致身体脂肪含量减少,瘦素合成分泌下降,通过减少与LEPR的结合而导致下丘脑神经细胞POMC表达下降;POMC分泌下降导致α-MSH 浓度降低,进而由于MC4R不被结合增加而产生食欲升高的生理效应。通过瘦素介导的食欲调节使人的体重在正常生理条件下可稳定在一定的范围内;当以上任何基因发生突变而导致上述反馈过程被打破时,人表现为嗜食和肥胖。
新发现的肥胖基因
Nature Genetics发表研究报告,解析6个新发现的基因,这6个新发现的基因通过影响神经中枢控制肥胖。研究小组通过全基因组关联分析法对32000个人进行分析,筛选出大量的与肥胖相关的变异,并获得6个新的肥胖基因。这6个基因分别是:TMEM18, KCTD15, GNPDA2, SH2B1, MTCH2, 和NEGR1。基因表达分析发现,6个基因都是在脑细胞中具有活性,以前研究发现的两个基因FTO和MC4R也具有相似的特性。德国国立基因组研究中心的教授表示,将通过其他的途径进一步研究这6个基因的功能。
最新进展
最近,《JCI Insight》发布的一篇文章,美国国立卫生研究院(NIH)下属的尤尼斯•肯尼迪•施赖弗国家儿童健康和人类发展研究所(NICHD)的研究人员通过小鼠实验发现,大脑中一种被称为Prkar2a的基因或许是控制肥胖的关键,该基因可以调节小鼠对脂肪和含糖食物的渴望以及对运动的渴望。

DOI:10.1172/jci.insight.141670
cAMP依赖性蛋白激酶(PKA)信号转导是介导交感神经信号分子和激素调节能量代谢的主要途径之一,这种由四个亚基组成的四聚体在细胞中具有包括调节糖原、糖和脂质的代谢等多种功能。Prkar2a编码PKA调节亚基IIα(RIIα)。在先前的研究中,NICHD的研究人员发现,与正常小鼠相比,不表达Prkar2a的小鼠即使接受高脂肪饮食饲养也相对不容易肥胖。于是,在这项新研究中,研究人员对其中的机制进行了探索。具体而言,研究人员对Prkar2a在缰核(Hb)中的作用进行了分析,后者是位于丘脑背侧的一个体积较小的双侧核团,含多种不同类型的神经元。近年来积累的证据表明,该部位参与对疼痛、压力、焦虑、睡眠和奖励的反应。在这项研究中,研究人员发现,Prkar2a几乎仅在内侧缰核(MHb)中表达,并且该基因是 MHb内细胞亚群中唯一高表达且差异表达的PKA调节亚单位。
敲除了小鼠的RIIα基因使之不表达Prkar2a后,小鼠PKA酶活性显著降低,基础活性趋于减弱。进一步分析表明,Prkar2a缺失诱导的PKA信号失调会导致缰核以及脚间核(IPN,MHb的主要传出物)的乙酰胆碱(一种神经递质)释放减少,同时造成IPN的谷氨酸水平下降。为了确定不表达Prkar2a的小鼠是否在自由进食的情况下改变了饮食习惯,研究人员对RIIα KO和野生型小鼠进行了高脂饮食(HFD)、高糖饮料以及运动偏好。与野生小鼠相比,不表达Prkar2a的小鼠无论是在食物充足还是禁食的情况下,都表现出进食更少的食物,食用更少的含糖饮料,并且运动量是野生小鼠的2-3倍。另外,不表达Prkar2a雌性小鼠会比雄性小鼠消耗更少高脂肪食物,而后者会比前者摄入更少的糖分。总之,这项研究揭示了PKA-RIIα在MHb中的独特作用,为今后人们探索如何预防肥胖症及其伴随的心血管疾病和糖尿病风险提供了重要参考。







